Clinical research

The clinical and preclinical analytical unit includes Good Laboratory Practice (GLP) laboratories in certified for:
- pharmacokinetic studies,
- bioanalytical studies.
We specialize in the development, transfer and validation of bioanalytical methods for the assessment of pharmacokinetics, pharmacodynamics and immunogenicity of biosimilar drugs in accordance with current ICH guidelines and those of the major drug regulatory authorities: EMA and FDA.
We have developed sets of biological methods applicable to the analysis of samples from preclinical and clinical studies. These include three specialized groups of assays.
Pharmacokinetic tests
Test:
Evaluation of monoclonal antibody pharmacokinetics using high-throughput platforms.
Effect:
The use of a high-throughput and fully automated platform in pharmacokinetics assessment methods significantly increases the quality of the data obtained while reducing the analysis time compared to classical assays.
Pharmacodynamic testing
Test:
Assessment of pharmacodynamics using flow cytometry.
Effect:
Acceleration, automation, standardization and high control of the performed tests.
Immunogenicity studies
Test:
Immunogenicity assessment (“Screening”, “Confirmatory” and “Titer” assays) using high throughput platforms and detection of NAb neutralizing antibodies using ADCC cell-based assays.
Effect:
High speed, reproducibility and sensitivity of testing at a level consistent with the expectations of regulatory authorities.
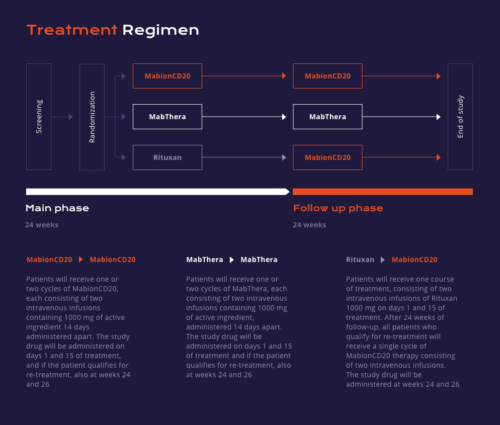
We are currently conducting a randomized, double-blind clinical trial MabionCD20-003RA (MABRIDGE) evaluating similarity between MabionCD20 and reference products (the EU-registered drug product MabThera and the US-registered drug product Rituxan).

Patients with moderate to severe rheumatoid arthritis diagnosed according to ACR 2010 criteria will be randomized to one of three groups in a 3:3:2 ratio.
The first group will receive MabionCD20, the second group will receive MabThera and the third group will receive Rituxan. Methotrexate will also be administered throughout the treatment period. The blinded drug will be given as two intravenous infusions (Main Phase) on days 1 and 15 of treatment.
Patients will then be followed up for at least 24 weeks to compare the pharmacokinetics, pharmacodynamics, efficacy, immunogenicity and safety between the three drug products.
Patients may receive a second cycle of treatment (Continuation Phase) at week 24, provided they meet the eligibility criteria for re-treatment as defined in the Clinical Trial Protocol. Groups treated with MabionCD20 and MabThera will continue their assigned treatment, while patients who received Rituxan in the Main Phase will take MabionCD20 in the Continuation Phase. All patients will be followed up until Week 48 in order for the safety, immunogenicity and efficacy data to be collected.
The MabionCD20-003RA clinical trial is conducted in accordance with the requirements defined in national legislation and international regulations. The proper conduct of the study is supervised by appointed authorities that evaluate the compliance of the study with the principles of ethics and ensure the protection of patients’ rights. An independent ethics committee and relevant regulatory authorities have issued positive opinions and approvals for the MabionCD20-003RA study.
The study is conducted by qualified personnel in specialized clinical centers, and its proper course is closely monitored.
Inclusion criteria:
Exclusion criteria:
-
- 1. Men or women between the ages of 18 and 80 years.
2. Body surface area (BSA) between 1.5 and 2.2 m².
3. Confirmed RA diagnosed according to the 2010 updated ACR/EULAR criteria. Duration of disease: at least 6 months prior to screening visit.
- 4. Moderate to severe RA despite treatment with methotrexate. Moderate to severe disease severity is defined as meeting the following criteria:
- 6 swollen joints and 6 tender/painful joints as determined by the study doctor at the screening visit and confirmed at visit 1;
- DAS28-ESR score of 3.2 at the screening visit.
- 5. No history of treatment with a TNF-α inhibitor (innovator or biosimilar, registered or investigational) prior to the screening visit.
6. Treatment with methotrexate at a dose of 7.5-25 mg per week for at least 12 weeks prior to the screening visit, with the dose remaining constant for the last 4 weeks; maintaining this dose throughout the study.
7. Consent to use highly effective contraceptive methods, from the screening visit until 12 months after the last dose of study drug (men, women of childbearing age).
8. Patients must not be pregnant or breastfeeding.
- Past or active inflammatory joint disease other than RA.
- History or active autoimmune disease.
- Class IV disease according to the ACR classification.
- Psychiatric disorders which could affect the normal course of the study.
- HBV, HCV, HIV infection.
- SARS-CoV-2 infection within 14 days prior to study drug administration and documented positive RT-PCR within 72 hours prior to first drug infusion or positive antigen test within 24 hours prior to first drug infusion.
- Serious and/or inadequately controlled comorbidities that are a contraindication to the administration of rituximab, methotrexate, or any of the drugs taken by patients during premedication, or that are high risk factors for the development of severe and life-threatening SARS-CoV-2 infection. Other diseases, in the opinion of the Investigator, also exclude the patient from the study. This category includes, but is not limited to, severe pulmonary, renal, hepatic, cardiovascular, neurological conditions, and severe and inadequately controlled type 1 or type 2 diabetes.
- Recent or active bacterial, viral, or fungal infection (excluding fungal nail bed infections).
- Active tuberculosis with typical symptoms of M. tuberculosis infection confirmed by positive screening results or history of tuberculosis (confirmed in the patient’s medical record, diagnosed prior to the screening period).
- Latent tuberculosis confirmed by the patient’s medical record or by the QuantiFERON test performed during the screening period in the absence of typical symptoms of the disease. A patient with latent TB may be eligible if he or she meets the following criteria:
- the patient has completed standard TB prophylaxis prior to the screening period and has no active TB or contact with an active case of TB after completion of TB prophylaxis or has received standard TB prophylaxis for at least 4 weeks prior to the screening visit and wishes to continue this regimen during the study;
- the patient does not have active tuberculosis at the time of screening, which must be confirmed by a pulmonologist, if at least 1 year has passed since completion of the last prophylaxis, or if the prophylaxis is being continued up to the time of screening;
- the patient has not had lesions detected on chest x-ray during the screening period and 3 months prior to the screening period.
- Malignancy within 5 years prior to the screening period (solid tumors, hematologic malignancies, and other cancers).
- History of severe cytopenia or other hematopoietic disorders.
- Primary or secondary immunodeficiency syndrome.
- Any other disorder that is listed as a contraindication to treatment with rituximab or methotrexate.
- Taking biologic or non-biologic DMARDs other than methotrexate at a specific time (specific guidelines defined in the Clinical Trial Protocol).
- Treatment with a TNF-α inhibitor (registered or investigational) prior to the screening period.
- History of treatment with B-lymphocyte modulating or B-lymphocyte reducing therapies, including rituximab and other anti-CD20 monoclonal antibodies (ocrelizumab, ofatumumab, obinutuzumab), belimumab, atacicept, tabalumab, epratuzumab and other experimental therapies.
- Use of systemic corticosteroids at a dose exceeding 10 mg prednisolone per day, or equivalent, within 2 weeks prior to the screening period or between the screening period and day 1 of therapy.
- Intraarticular injections of hyaluronic acid within 28 days prior to the screening period or between the screening period and day 1 of therapy.
- Use of any drug that has not been approved by a regulatory authority for any indication within 4 weeks or 5 half-lives (whichever is longer) prior to the screening visit or planned administration of a drug/vaccine not approved by a regulatory authority during the study period.
- History of anaphylactic reaction related to treatment with rituximab or any excipient contained in the study drug.
- Abnormal laboratory findings, specifically:
- White blood cell count <3,000/μL or neutrophil count <1,500/μL.
- Platelet count <75,000/μL.
- Aspartate aminotransferase or alanine aminotransferase >2.5 x ULN.
- Hemoglobin <8.0 g/dL.
- IgG <5.0 mg/mL or IgM <0.4 mg/mL.
- Other abnormal laboratory findings (clinically significant).
- Intolerance or contraindications to methotrexate, drugs included in premedication or intravenous corticosteroids.
- Major surgery (including joint surgery) within 8 weeks prior to the screening visit or planned surgery within 12 months after starting treatment.
- Recent vaccination with an inactivated/live vaccine (<4 weeks prior to 1st dose of drug) or live vaccine (6 weeks prior to 1st dose of drug).
- Planned vaccination with a live vaccine during the study period.
- Chronic use of narcotic analgesics (morphine, fentanyl, hydrocodone, oxycodone, codeine).
- Participation in another clinical trial within 2 months prior to study entry (exception: – screen failure in the MabionCD20-003RA trial).
- Patients who are breastfeeding, pregnant or planning a pregnancy within 12 months of the last administration of study drug.
- Blood donation or loss of more than 500 mL of blood within 2 months prior to the screening visit.
- Lack of access to peripheral veins.
- Drug, alcohol, substance abuse within 2 years prior to the screening visit.