Badania kliniczne

W skład jednostki analityki klinicznej i przedklinicznej wchodzą laboratoria działające w systemie Dobrej Praktyki Laboratoryjnej (GLP), certyfikowane w zakresach:
- badania farmakokinetyczne,
- badania bioanalityczne.
Specjalizujemy się w rozwoju, transferze oraz walidacji metod bioanalitycznych oceny farmakokinetyki, farmakodynamiki oraz immunogenności leków biopodobnych – zgodnie z obowiązującymi wytycznymi ICH oraz głównych organów rejestrujących leki: EMA i FDA.
Rozwinęliśmy zestawy metod biologicznych mających zastosowanie w analizie prób z badań przedklinicznych oraz klinicznych.
Obejmują one trzy specjalistyczne grupy testów, realizowanych zgodnie ze standardami GCP oraz GLP.
Badania farmakokinetyczne
Test:
Ocena farmakokinetyki przeciwciał monoklonalnych z wykorzystaniem platform wysokoprzepustowych.
Efekt:
Zastosowanie wysokoprzepustowej i w pełni automatycznej platformy w metodach oceny farmakokinetyki w znacznym stopniu zwiększające jakość uzyskiwanych danych przy jednoczesnym skróceniu czasu wykonania analizy w porównaniu do testów klasycznych.
Badania farmakodynamiczne
Test:
Ocena farmakodynamiki dostosowana do mechanizmu akcji badanego terapeutyku z wykorzystaniem wyselekcjonowanych technik analitycznych, w tym cytometrii przepływowej, zautomatyzowanych systemów do analiz immunoenzymatycznych i innych.
Efekt:
Przyspieszenie, automatyzacja, standaryzacja i wysoka kontrola realizowanych badań.
Badania immunogenności
Test:
Ocena immunogenności (testy „Screening”, „Confirmatory” oraz „Titer”) z wykorzystaniem platform wysokoprzepustowych oraz wykrywanie przeciwciał neutralizujących NAb z wykorzystaniem testów komórkowych ADCC.
Efekt:
Wysoka czułość, szybkość i powtarzalność testowania na poziomie zgodnym z oczekiwaniami organów regulatorowych
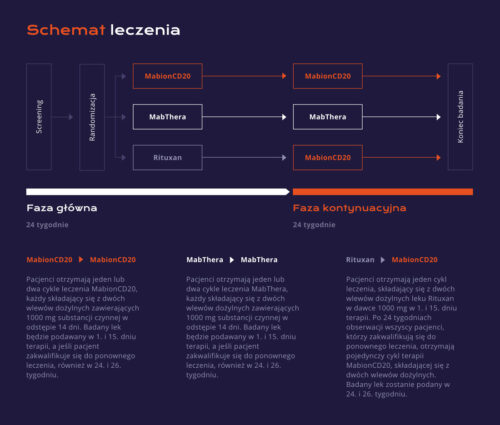
Obecnie prowadzimy randomizowane, podwójnie zaślepione badanie kliniczne MabionCD20-003RA (MABRIDGE), porównujące farmakokinetykę oraz kliniczne podobieństwo między MabionCD20 i produktami referencyjnymi (zarejestrowanym w UE produktem leczniczym MabThera i zarejestrowanym w USA produktem leczniczym Rituxan).

Pacjenci z reumatoidalnym zapaleniem stawów o nasileniu od umiarkowanego do ciężkiego, zdiagnozowani zgodnie z kryteriami ACR 2010, zostaną przydzieleni losowo do jednej z trzech grup w stosunku 3:3:2.
Pierwsza grupa będzie leczona preparatem MabionCD20, druga grupa otrzyma preparat MabThera, a trzecia – preparat Rituxan. Przez cały okres leczenia będzie podawany także metotreksat. Zaślepiony lek zostanie podany w dwóch wlewach dożylnych (Faza Główna) w 1 i 15 dniu terapii. Pacjenci będą następnie pod obserwacją lekarską przez minimum 24 tygodnie w celu oceny i porównania farmakokinetyki, efektywności, farmakodynamiki i bezpieczeństwa między trzema produktami leczniczymi.
Pacjenci mogą otrzymać drugi cykl terapii (Faza Kontynuacyjna) w 24. tygodniu pod warunkiem, że będą spełniać kryteria kwalifikujące do ponownego leczenia, określone w Protokole Badania Klinicznego. Grupy leczone MabionCD20 i preparatem MabThera będą kontynuować przydzielone im terapie, natomiast pacjenci, którzy otrzymają Rituxan w Fazie Głównej, w Fazie Kontynuacyjnej będą przyjmować MabionCD20. Wszyscy pacjenci będą pod obserwacją lekarską aż do 48. tygodnia w celu zebrania danych dotyczących bezpieczeństwa, immunogenności i efektywności terapii.
Badanie kliniczne MabionCD20-003RA jest prowadzone zgodnie z wymaganiami zdefiniowanymi w krajowych przepisach prawnych i regulacjach międzynarodowych. Nad jego prawidłowym przebiegiem czuwają wyznaczone do tego instytucje, które oceniają zgodność badania z zasadami etyki oraz dbają o ochronę praw pacjentów. Niezależna komisja bioetyczna oraz właściwe organy regulacyjne wydały pozytywne opinie i zgody dotyczące badania MabionCD20-003RA.
Badanie jest prowadzone przez wykwalifikowany personel w specjalistycznych ośrodkach klinicznych, a jego właściwy przebieg jest ściśle monitorowany.
Kryteria włączenia:
Kryteria wyłączenia:
1. Mężczyźni lub kobiety w wieku 18-80 lat.
2. Powierzchnia ciała (BSA) między 1.5 a 2.2 m².
3. Potwierdzone RZS, zdiagnozowane zgodnie ze zaktualizowanymi w 2010 r. kryteriami ACR/EULAR. Czas trwania choroby: co najmniej 6 miesięcy przed wizytą przesiewową.
4. RZS o nasileniu od umiarkowanego do ciężkiego, pomimo leczenia metotreksatem. Umiarkowane lub ciężkie nasilenie choroby definiuje się jako spełnienie następujących kryteriów:
- 6 obrzękniętych stawów i 6 tkliwych/bolesnych stawów, stwierdzonych przez lekarza podczas wizyty przesiewowej i potwierdzonych podczas wizyty 1.;
- wynik DAS28-ESR 3.2 podczas wizyty przesiewowej.
5. Brak historii leczenia inhibitorem TNF-α (innowacyjnym lub biopodobnym, zarejestrowanym lub badanym) przed wizytą przesiewową.
6. Leczenie metotreksatem w dawce 7.5-25 mg na tydzień przynajmniej przez okres 12 tygodni przed wizytą przesiewową, przy czym dawka musi być stała przez ostatnie 4 tygodnie; przyjmowanie tej dawki przez cały okres trwania badania.
7. Zgoda na stosowanie wysokoefektywnych metod antykoncepcyjnych, od wizyty przesiewowej aż do 12 miesięcy po przyjęciu ostatniej dawki leku badanego (mężczyźni, kobiety w wieku rozrodczym).
8. Pacjentki nie mogą być w ciąży ani karmić piersią.
1. Przebyta lub aktywna choroba zapalna stawów inna niż RZS.
2. Przebyte lub aktywne choroby autoimmunologiczne.
3. IV klasa choroby wg klasyfikacji ACR.
4. Zaburzenia psychiczne, które mogłyby wpłynąć na prawidłowy przebieg badania.
5. Zakażenie wirusami HBV, HCV, HIV.
6. Zakażenie wirusem SARS-CoV-2 w ciągu 14 dni przed podaniem badanego leku oraz udokumentowany pozytywny wynik testu RT-PCR w ciągu 72 godz. przed pierwszą infuzją leku lub pozytywny wynik testu antygenowego w ciągu 24 godz. przed pierwszą infuzją leku.
7. Poważne i/lub nieodpowiednio kontrolowane choroby współistniejące, które są przeciwskazaniem do podawania rytuksymabu, metotreksatu lub któregokolwiek z leków przyjmowanych przez pacjentów w czasie premedykacji bądź będące czynnikami wysokiego ryzyka rozwoju ciężkiej i zagrażającej życiu infekcji SARS-CoV-2. Również inne choroby, które zgodnie z opinią Badacza wykluczają pacjenta z badania. Do tej kategorii należą m.in. ciężkie choroby płuc, nerek, wątroby, układu krążenia, neurologiczne, ciężka i nieodpowiednio kontrolowana cukrzyca typu 1 lub 2.
8. Niedawno przebyta lub aktywna infekcja bakteryjna, wirusowa lub grzybicza (wyłączając grzybicze infekcje łożysk paznokciowych).
9. Aktywna gruźlica z typowymi objawami zakażenia M.tuberculosis potwierdzonymi dodatnimi wynikami badania przesiewowego lub przebyta gruźlica (potwierdzona w dokumentacji medycznej pacjenta, zdiagnozowana przed okresem przesiewowym).
10. Gruźlica utajona potwierdzona w dokumentacji medycznej pacjenta lub za pomocą testu QuantiFERON przeprowadzanego w czasie okresu przesiewowego, przy braku typowych objawów choroby. Pacjent z gruźlicą utajoną może kwalifikować się do badania, jeśli spełnia następujące kryteria:
- pacjent ukończył standardową profilaktykę gruźlicy przed okresem przesiewowym i nie miał aktywnej gruźlicy ani nie miał kontaktu z aktywnym przypadkiem gruźlicy po zakończeniu profilaktyki gruźlicy lub otrzymywał standardową profilaktykę gruźlicy przez co najmniej 4 tygodnie przed wizytą przesiewową i chce kontynuować ten schemat podczas badania;
- pacjent nie ma aktywnej postaci gruźlicy w czasie badania przesiewowego, co musi zostać potwierdzone przez pulmonologa, jeśli od zakończenia ostatniej profilaktyki minął co najmniej rok lub do czasu badania przesiewowego profilaktyka jest nadal kontynuowana;
- u pacjenta nie zostały wykryte zmiany podczas prześwietlenia klatki piersiowej w czasie okresu przesiewowego i 3 miesiące przed okresem przesiewowym.
11. Choroba nowotworowa w ciągu 5 lat przed okresem przesiewowym (guzy lite, nowotwory hematologiczne i inne).
12. Przebyta ciężka cytopenia lub inne zaburzenia układu krwiotwórczego.
13. Pierwotny lub wtórny niedobór odporności.
14. Każde inne zaburzenie, które jest wymienione jako przeciwwskazanie do leczenia rytuksymabem lub metotreksatem.
15. Przyjmowanie biologicznych lub niebiologicznych leków DMARD innych niż metotreksat w określonym czasie (szczegółowe wytyczne zdefiniowane w Protokole Badania Klinicznego).
16. Leczenie inhibitorem TNF-α (zarejestrowanym lub badanym) przed okresem przesiewowym.
17. Przebyte leczenie za pomocą terapii modulującej limfocyty B lub zmniejszającej ilość limfocytów B, m.in. rytuksymabem i innymi przeciwciałami monoklonalnymi anty-CD20 (okrelizumab, ofatumumab, obinutuzumab), belimumab, atacicept, tabalumab, epratuzumab i innymi terapiami eksperymentalnymi.
18. Stosowanie ogólnoustrojowych glikokortykosteroidów w dawce większej niż 10 mg prednizolonu na dobę lub równoważnej, w ciągu 2 tygodni przed okresem przesiewowym lub między okresem przesiewowym a 1. dniem terapii.
19. Dostawowe iniekcje kwasu hialuronowego w ciągu 28 dni przed okresem przesiewowym lub między okresem przesiewowym i 1. dniem. terapii.
20. Stosowanie jakiegokolwiek leku, który nie został zatwierdzony przez organ regulatorowy w żadnym wskazaniu w ciągu 4 tygodni lub 5 okresów półtrwania (w zależności od tego, który jest dłuższy) przed wizytą przesiewową lub planowane przyjęcie leku/szczepionki niezatwierdzonego przez organ regulatorowy w czasie trwania badania.
21. Przebyta reakcja anafilaktyczna związana z leczeniem rytuksymabem lub jakąkolwiek substancją pomocniczą zawartą w badanym leku.
22. Nieprawidłowe wyniki badań laboratoryjnych, w szczególności:
- Liczba białych krwinek <3,000/μL lub liczba neutrofili <1,500/μL.
- Liczba płytek krwi <75,000/μL.
- Aminotransferaza asparaginianowa lub aminotransferaza alaninowa >2.5 x ULN.
- Hemoglobina <8.0 g/dL.
- IgG poniżej 5.0 mg/mL lub IgM poniżej 0.4 mg/mL.
- Inne nieprawidłowe wyniki badań laboratoryjnych (istotne klinicznie).
23. Nietolerancja lub przeciwwskazania do podawania metotreksatu, leków wchodzących w skład premedykacji lub dożylnie glikokortykosteroidów.
24. Poważna operacja (włączając operacje stawów) w ciągu 8 tygodni przed wizytą przesiewową lub planowana operacja w ciągu 12 miesięcy po rozpoczęciu leczenia.
25. Niedawne szczepienie szczepionką inaktywowaną/nieżywą (<4 tygodni przed podaniem 1. dawki leku) lub szczepionką żywą (6 tygodni przed podaniem 1. dawki leku).
26. Planowane szczepienie żywą szczepionką w czasie trwania badania.
27. Przewlekłe przyjmowanie narkotycznych leków przeciwbólowych (morfina, fentanyl, hydrokodon, oksykodon, kodeina).
28. Udział w innym badaniu klinicznym w ciągu 2 miesięcy przed włączeniem do badania (wyjątek – nieudane procedury przesiewowe w badaniu MabionCD20-003RA).
29. Pacjentki karmiące piersią, w ciąży lub planujące ciążę w ciągu 12 miesięcy od ostatniego podania leku badanego.
30. Oddanie krwi lub utrata powyżej 500 ml krwi w ciągu 2 miesięcy przed wizytą przesiewową.
31. Brak dostępu do żył obwodowych.
32. Nadużywanie narkotyków, alkoholu, substancji chemicznych w ciągu 2 lat przed wizytą przesiewową.